Protein Imputation#
[ ]:
%%bash
git clone https://github.com/Zafar-Lab/multiHIVE.git
pip install scvi-tools
pip install scanpy
pip install anndata
pip install scikit-misc
Cloning into 'multiHIVE'...
remote: Enumerating objects: 49, done.
remote: Counting objects: 100% (49/49), done.
remote: Compressing objects: 100% (39/39), done.
remote: Total 49 (delta 9), reused 44 (delta 7), pack-reused 0 (from 0)
Receiving objects: 100% (49/49), 18.85 KiB | 2.69 MiB/s, done.
Resolving deltas: 100% (9/9), done.
[4]:
import scvi
import scanpy as sc
import sys
import numpy as np
import matplotlib.pyplot as plt
sys.path.append("/content/multiHIVE")
from src.model import HierarVI
INFO:lightning_fabric.utilities.seed:Global seed set to 0
[5]:
adata = scvi.data.pbmcs_10x_cite_seq()
INFO Downloading file at data/pbmc_10k_protein_v3.h5ad
Downloading...: 24938it [00:00, 92329.13it/s]
INFO Downloading file at data/pbmc_5k_protein_v3.h5ad
Downloading...: 100%|██████████| 18295/18295.0 [00:00<00:00, 68158.18it/s]
/usr/local/lib/python3.11/dist-packages/scvi/data/_built_in_data/_cite_seq.py:48: ImplicitModificationWarning: Setting element `.obsm['protein_expression']` of view, initializing view as actual.
dataset1.obsm["protein_expression"] = pd.DataFrame(
/usr/local/lib/python3.11/dist-packages/scvi/data/_built_in_data/_cite_seq.py:53: ImplicitModificationWarning: Setting element `.obsm['protein_expression']` of view, initializing view as actual.
dataset2.obsm["protein_expression"] = pd.DataFrame(
/usr/local/lib/python3.11/dist-packages/anndata/_core/anndata.py:1818: UserWarning: Observation names are not unique. To make them unique, call `.obs_names_make_unique`.
utils.warn_names_duplicates("obs")
[6]:
adata
[6]:
AnnData object with n_obs × n_vars = 10849 × 15792
obs: 'n_genes', 'percent_mito', 'n_counts', 'batch'
obsm: 'protein_expression'
[7]:
adata.layers["counts"] = adata.X.copy()
sc.pp.normalize_total(adata)
sc.pp.log1p(adata)
adata.raw = adata
adata.obs_names_make_unique()
[8]:
#will automatically detect proteins as missing in a certain batch if the protein has 0 counts
# for each cell in the batch. In other words, to indicate a protein is missing in a certain batch,
# please set it to 0 for each cell.
batch = adata.obs.batch.values.ravel()
held_out_proteins = adata.obsm["protein_expression"][batch == "PBMC5k"].copy()
adata.obsm["protein_expression"].loc[batch == "PBMC5k"] = np.zeros_like(
adata.obsm["protein_expression"][batch == "PBMC5k"]
)
[9]:
sc.pp.highly_variable_genes(
adata,
n_top_genes=4000,
flavor="seurat_v3",
batch_key="batch",
subset=True,
layer="counts"
)
HierarVI.setup_anndata(
adata,
layer="counts",
batch_key="batch",
protein_expression_obsm_key="protein_expression"
)
<frozen abc>:119: FutureWarning: SparseDataset is deprecated and will be removed in late 2024. It has been replaced by the public classes CSRDataset and CSCDataset.
For instance checks, use `isinstance(X, (anndata.experimental.CSRDataset, anndata.experimental.CSCDataset))` instead.
For creation, use `anndata.experimental.sparse_dataset(X)` instead.
INFO Using column names from columns of adata.obsm['protein_expression']
INFO Found batches with missing protein expression
<frozen abc>:119: FutureWarning: SparseDataset is deprecated and will be removed in late 2024. It has been replaced by the public classes CSRDataset and CSCDataset.
For instance checks, use `isinstance(X, (anndata.experimental.CSRDataset, anndata.experimental.CSCDataset))` instead.
For creation, use `anndata.experimental.sparse_dataset(X)` instead.
[13]:
import numpy as np
import matplotlib.pyplot as plt
vae = HierarVI(adata)
vae.train(max_epochs=200)
adata.obsm["Zs1"], adata.obsm["Zs2"], adata.obsm["Zr"], adata.obsm["Zp"] = vae.get_latent_representation()
adata.obsm['Z_multiHIVE'] = np.concatenate((adata.obsm["Zs1"], adata.obsm["Zr"], adata.obsm["Zp"]), axis=1)
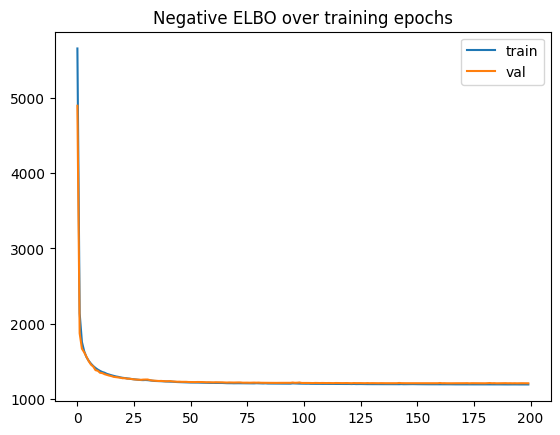
plt.plot(vae.history["elbo_train"], label="train")
plt.plot(vae.history["elbo_validation"], label="val")
plt.title("Negative ELBO over training epochs")
plt.legend()
[13]:
<matplotlib.legend.Legend at 0x7b15ca79acd0>
[12]:
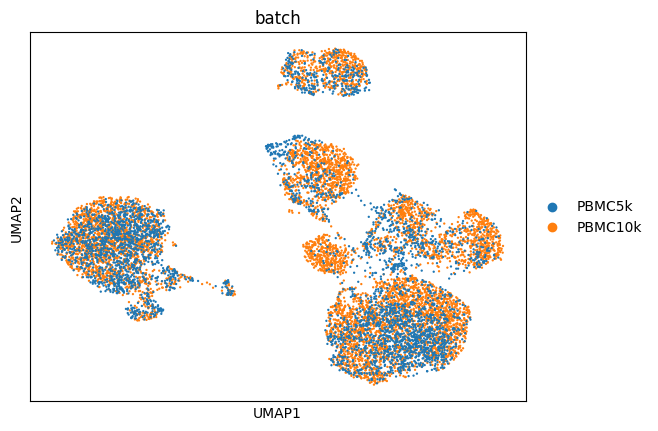
sc.pp.neighbors(adata, use_rep = "Z_multiHIVE")
sc.tl.umap(adata)
sc.pl.umap(adata, color = "batch")
[15]:
_, protein_means = vae.get_normalized_expression(
n_samples=25,
transform_batch="PBMC10k",
include_protein_background=True,
sample_protein_mixing=False,
return_mean=True,
)
<frozen abc>:119: FutureWarning: SparseDataset is deprecated and will be removed in late 2024. It has been replaced by the public classes CSRDataset and CSCDataset.
For instance checks, use `isinstance(X, (anndata.experimental.CSRDataset, anndata.experimental.CSCDataset))` instead.
For creation, use `anndata.experimental.sparse_dataset(X)` instead.